Protocols - SOPs
Analytical Gel Protocol for Polyacrylamide Gel Electrophoresis (PAGE)
1. Prep 10% APS
a. Weigh 100mg of APS into 2mL tube
b. Add 1mL of DI water
c. This should be dissolved by the time it is needed
2. Clean Gel Plates
a. Rinse first with DI, dry with large Kim Wipes
b. Then rinse with EtOH, dry with large wipes
c. Coat plates with sigmacote–>only side that gel contacts (1-side)
i. Squirt (using pipet) 200µL of sigmacote on each plate + spread around using large wipe
ii. Purpose is to polish plates allowing gels to pour evenly (avoids bubbles in gel)
iii. Use glare from light to remove any smears + dust
iv. Plates only have to stay in hood until vapors stop (appears dry)
3. Clean Gasket, Spacers, & Comb
a. Rinse with DI water + wipe dry with Kim Wipes
4. Assemble + Level the Plates
a. Lay out several large Kim Wipes under plates to absorb any spills
b. Keep polished sides towards center/gel
c. Carefully wrap rubber gasket around the plate with the curved bottom and no cutout
d. Place the spacers next to the gasket on each side of the plate
e. Place the plate with the cutout on top of the first, making sure the gasket is sealed
f. Use small clips to secure the plates together and balance the plates upright
g. Spacers should not be able to move

5. Prep Gel Solution: 17% acrylamide by volume gel
a. Clean 250mL beaker rigorously: good practice to dedicate 1 beaker for gel prep only that is kept very clean
b. Keep beaker covered with parafilm when not in use
c. For 100mL of gel: (listed in order that they are added to beaker)
· Urea 42.02g (Keeps DNA denatured)
· 5xTBE 20mL (Maintains pH)
· Acrylamide/BisAcryl.(40%) 42.5mL (Polymerase gel) (nature.com/nprot/info/tools.html)
· H2O until total volume is 100mL
For 50mL of gel:
· Urea 21.01g
· 10xTBE 5mL
· Acryl./BisAcryl. 21.25mL
· H2O until total volume is 50mL
For non-denaturing gel, do not add Urea.
Volume Acryl/BisAcryl.: (percent acrylamide gel wanted * total volume being prepared) / (percent Acryl./BisAcryl. prepared)
(For example, here: 21.25mL=(17*50)/(40))
d. Next, nuke in microwave for 5 sec. to heat to RT as wanted for reaction because Acryl. was stored in fridge. Be careful not to overheat.
e. Mix gel using magnetic stirrer (set to 6 out of 10 on stirrer) for ~5 min (once all salt is in solutionàappears clear)
6. Finish Preparing Gel + Pour into Plates
a. First, use a 200-1000 µL + 20-100 µL pipet to extract: (2x volume if making 100mL of gel)
i. 32.5 µL concentrated TEMED
ii. 325 µL 10% weight/volume APS (Ammonium per Sulfate)
b. Bring pipets with chemicals to a pipet rack placed near beaker containing gel solutionàcareful not to contaminate tips!
c. While gel is still being stirred, lift parafilm + add TEMED+APS simultaneously
d. Allow to stir for about 10 seconds
e. Extract about 35mL (determined by volume between plates) of gel solution using 60mL syringe
f. Place tip of syringe in center of cutout in plate with tip touching glass + placed as close to edge as possible
g. Slowly squirt gel into plates
i. Gel should spread evenly + uniformly through plates if plates are cleaned + coated/polished properly
ii. Some gel will spill out near the syringe but not a problem
iii. Surface tension will cause gel to spread in plates rather than spill
h. Once plates appear filled, double-check that gel is flush with all edges of plates + make sure there are no bubbles
i. Allow to spill out around cutout slightly then stop
j. Then, place comb between plates on cutout making sure that wells are large enough for your sample
i. Check to make sure the insertion of comb did not cause gel to spill out end creating bubbles near far edge
k. Immediately after pouring gel, pour remaining contents of syringe back into beaker + recover beaker with parafilm with a very good seal. Rinse syringe in sink.
i. Beaker of remaining (unused) gel will be used to indicate when the gel has solidified between plates
l. Allow gel to sit 30-60 minutes (until it has solidified)
i. Change gloves immediately afteràassume monomers of acrylamide are present in solution making it harmful to touch
ii. When gel solidifies, it will shrink away from edge slightly-OK
7. Prepare Samples for PAGE
a. If tubes are completely dry (all EtOH removed), resuspend DNA in 15µL predyes
i. 0.1xTBE, 7M urea, 0.01% BPBàstain/loading dye for loading gels (Bromophenol Blue) (Can also use 5µL 50% glycerol)
b. Place tubes in heat rack + heat for 5 minutes at 95C
i. This step is only necessary if you’re performing the experiment the same day. If you just let solution sit overnight then DNA will resuspend on its own.
8. Rinse Gel Apparatus with DI + Dry with Kim Wipes
9. Remove Comb, Load Plates + fill wells with 1xTBE
a. Fill bottom well of apparatus with 1xTBE
b. Once wells of plates are empty, remove gasket and pick up plates + set in well (With cutout facing well)
i. Insert one corner of plates into bottom well on bumper first + then slowly tilt plates into well
ii. This will help avoid bubbles forming on the bottom of the plates</.p>
c. Use syringe with curved needle tip to remove bubbles that form along bottom edge of plate
d. Clip plates to apparatus
e. Fill top well with 1xTBE to about 1cm above wells
f. Slowly + gently remove comb from solidified gel + peel off gel from spillage
g. 1xTBE should flow into wells
10. Prepare Wells for DNA
a. Want to use wells in the center because that is where the most heat is generatedàincreases uniformity of DNA running in gels
b. Use a sharpie to number lanes 1-7 with wells on either side of 1-7 labeled X
c. X wells will be used during prerun of dyes
d. Remove any remaining urea in wells by removing some 1xTBE from reservoir with untipped syringe, then after placing straight needle tip on syringe squirt the solution into wells by inserting needle tip into bottom of well. Repeat 3 times for each well.
i. At first, you will see liquid swirl after rinsing due to the urea settling on the bottom (urea is heavier than 1xTBE)
ii. Swirling will not occur once urea has been sufficiently removed
e. Empty heat block, turn heat block to 96C to allow to begin heating during prerun (used in step 12)
11. Pre-Running Dyes to Ensure Gel Was Made Properly [Optional]
a. Put running dye in a PCR tube
i. Mix is made of 0.01% BPB + xylene cyanol
ii. Recommend buying pre-mixed in aqueous solution
b. Add 15 µL of the running dye to the two X labelled wells using pipet with gel tip
i. Keep tip at very bottom of well when adding the dyes
c. Attach leads of power source to gel apparatus (negative to top, positive to bottom)
d. Cover wells with glass covers
e. Set power to 27W + turn on for about 10 minutes
i. Be very careful not to touch the apparatus, particularly wells when power source is turned on. Current can seriously harm you!
ii. Running dyes will be pulled into gel
iii. The two dyes will separate because they behave like different lengths of DNA + create bands of color at different heights
Concentrations of Acrylamide giving optimum resolution of DNA fragments using denaturing PAGE (slightly varies by size of gel):
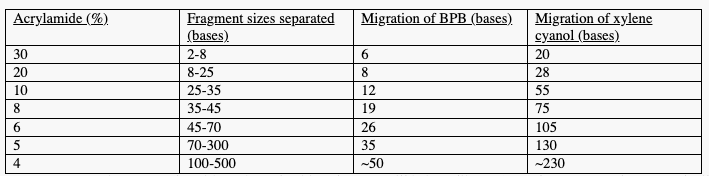
iv. Example: with 17% acrylamide gels, BPB will behave like an 8 mer fragment + xylene cyanol will behave like a 28 mer fragment
v. Shorter fragments are carried further through gel so xylene cyanol creates a teal blue band near top of gel while BPB will create a dark blue band further down the gel
f. Turn off power supply after 10 minutes
g. Look at bands to see if gel was made correctly:
Ideal Banding Acceptable Banding![]()
![]()
Unacceptable Banding
![]()
h. If dyes behave acceptably, we are now ready to run the actual samples!
12. Denature Samples 1-7
a. Heat block should be heated to 96C
b. Place tubes in heat block + allow to heat for 2-4 minutes to ensure all DNA is denatured
13. Load Samples into Wells
a. Using Pipet with gel tip, remove all liquid from each tube + load into corresponding well
i. Make sure tip is at the bottom of well while loading
ii. This should be done quickly, carefully + without any distractions
14. Turn On Power Supply
a. Set power to 200-250 V + allow gel to run until BPB reaches the bottom (about 90 minutes)
b. If available, clip a metal sheet onto outside of plate (WHEN POWER IS OFF)
i. Metal sheet will draw heat from the plates, protecting the plates +keep gel from melting
15. Turn Off Power + Empty Top Well
16. Remove + Image Gel
a. Remove plates from gel apparatus
b. Wash imaging tray using DI water, then EtOH, then dry with wipe
i. Imaging/gel tray is made of a special kind of plastic that allows UV rays to pass through
c. Carefully remove gasket, remove spacers separating plates by pulling away from plates
d. Use a pry bar to pull top plate off
i. Plate with cutout is on bottom
ii. Insert flat side between plates + gently turn to pry plate off
iii. Take top plate off
e. Slice 1 corner of gel from top to mark numbering
f. Slice off “combs” from top of gel using pry bar
g. Pick up corners of gel + gently lift gel off of plate
h. Lay gel on gel tray carefully + slowly, avoiding the formation of bubbles
i. Pull/rearrange gel to remove any bubbles that may have formed
j. Take gel tray to gel-dock for imagingàGel Doc-It Imaging System with UVP
i. Gel dock shines UV-light from bottom + takes an image of fluorescing bands with camera
17. Stain Gel to Image All Bands
a. Dilute DNA Stain:
i. Add 150 µL of 1xTBE to mixing dish/tub
ii. Add 15 µL of dyeàin this case sybr gold
iii. Mix with tip of pipet until appears uniform
b. Pour about ¾ of diluted stain into gel staining tray
c. Place gel into tray avoiding formation of bubbles
d. Pour remaining diluted stain onto gel
i. Make sure gel is completely submerged in liquid
e. Stain for about 10 minutes, agitating the tray (moving around) every 3 minutes
i. Cover tray with foil during staining to prevent photobleaching of dye
f. Pour out dye and wash with DI, agitating the tray
g. Clean gel imaging tray with DI water + EtOH
h. Transfer gel to imaging tray, avoiding bubbles
i. Take tray with gel to gel dock and image gel